As electricity from renewable sources increases in scale and decreases in price, the need for electrochemical energy conversion is growing. Renewable electricity can be used to drive water electrolysis, producing hydrogen, or to drive CO2 electrolysis, producing carbon-based renewable liquid fuels. Either way, the cathode with the fuel-producing half-reaction has to be balanced by an anode oxidizing water to O2. The thermodynamic potential for oxidizing water is 1.23 V versus the reversible hydrogen electrode, but in practice water-oxidizing anodes run at a significantly higher potential. This difference in potential, the overpotential, is the main factor limiting the efficiency of electrolysis cells, and so there’s a lot to gain in improving water oxidation catalysis.
Nickel-iron-based electrodes can drive oxidation of water to O2 with the least overpotential of any known material in alkaline environment, and are therefore used in alkaline water electrolysers. However, a number of fundamental questions remain unanswered. One of these questions, which we considered the top priority, is whether or not the catalytic activity is confined to the surface of the material. Only with an answer to this question is it possible to compare the intrinsic activity of electrocatalyst materials, which is most fundamentally defined as the rate at which a single site produces O2 molecules, a quantity known as the turn-over frequency. Determining the turn-over frequency is an essential starting point for rational design of materials for more efficient electrolyser anodes.
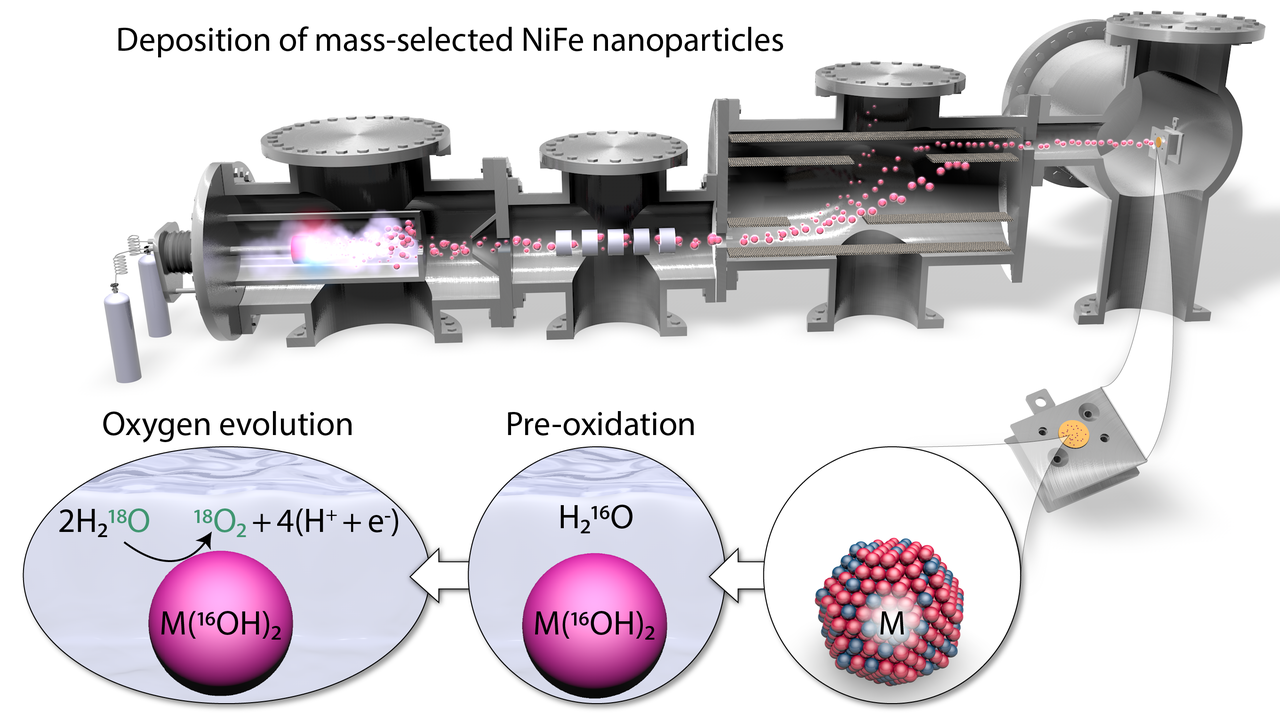
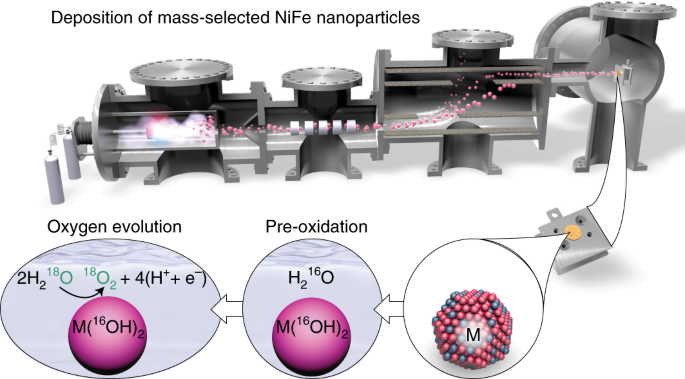
In attempting to answer the question of whether activity is confined to the outer surface, we realized that the concept of surface is not very well-defined for the electrodeposited nickel-iron-based electrodes typically studied. They are highly porous and permeable to the electrolyte, which is good for achieving high current densities in industrial electrolysers, but bad for answering fundamental questions. So we synthesized a model system: spherical nanoparticles of 75% nickel and 25% iron with a narrow mass distribution and finely controlled loading, formed and deposited in ultra-high vacuum. Since we knew the total outer surface area, we were able to determine that the nickel redox feature usually used as a measure of the catalytically active surface actually probes a near-surface region approximately three atomic layers thick. We also showed that the catalytic activity correlates with the surface area but not with the total loading, excluding the possibility of an electrocatalytically active bulk.
Whether the catalytic activity is limited to the outer surface or extends into the near-surface region is a more difficult question to answer. If there were activity below the outer surface, we reasoned, that would probably mean that some of the oxygen evolved would come from the nanoparticles themselves and not directly from water. If that was happening, we reasoned, then we could detect it by isotope labeling. This argument depends on any oxygen in the near-surface region being immobile when a potential is not applied, but given the compact appearance of the nanoparticles in transmission electron microscopy images, we think that this is a reasonable assumption. So we prepared nanoparticle samples in electrolyte made from normal 16-O water, and then used them to oxidize isotope-labeled 18-O water while measuring the O2 produced with a mass spectrometer. We found that less than one percent of one atomic layer’s worth of oxygen, if any, came from the catalyst and not from the water. We got the same result when we switched the order of the two isotopes. What’s more, using low-energy ion scattering spectroscopy after the electrochemistry experiment, we saw that the first isotope of oxygen was always still on the surface as well as immediately below the surface. While we can’t completely rule out high mobility of oxygen species in the near-surface region, we think that a lack of subsurface activity is a more likely explanation.
Armed with this evidence that activity is limited to the outer surface, we were able to report a new lower limit for the turn-over frequency of the active sites: 6 molecules of oxygen per surface metal atom per second at an overpotential of 300 mV. This is higher than all previous estimates of turn-over frequency for oxygen evolution in alkaline environment. Incidentally, our isotope result also helps to answer another question about this electrocatalyst. Since the surface-terminating lattice oxygen is also of a different isotope than the water being oxidized, the lack of an isotope exchange signal means that lattice oxygen is not involved in the electrocatalytic cycle. This supports an associative mechanism involving a single adsorption site and rules out several other proposed mechanisms.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in