
One of the main research areas in the Glorius group is addressing challenges in the asymmetric hydrogenation of (hetero)arenes. We mainly focused on using ruthenium-carbene complexes, which enable the formation of saturated heterocycles in a highly stereoselective manner. Over time, we became aware of different synthetic problems associated with arene hydrogenation. Chiefly among these is chemoselectivity, e.g. hydrogenation of the aromatic ring without losing the directly-attached functional groups owing to further hydrogenation or hydrodefunctionalization.
Along these lines, and being aware of the possible obstacles, we were motivated to approach the hydrogenation of fluoroarenes towards the generation of all-cis-(multi)fluorinated cycloalkanes. Our successful protocol, which was recently published in Science, enabled the preparation of a library of fluorinated aliphatic motifs in high yields and diastereoselectivities. It also allowed access to multifluorinated cycloalkanes in a single synthetic step, including the all-cis-hexafluorocyclohexane, which was previously obtained via a 12-step synthesis reported in Nature Chem. by the O'Hagan group.
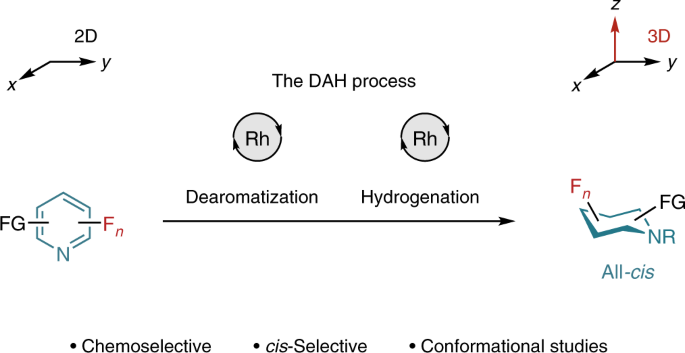
However, one important piece of the puzzle was missing! The formation of fluorinated piperidines was not possible at that point. We tried to tackle this problem by looking for different reaction conditions, including alternative catalysts. Unfortunately, we were unable to reduce pyridine precursors owing to, we believe, catalyst poisoning by the substrate or the product. In some cases, we observed trace reactivity, but hydrodefluorinated products were the main outcome. We therefore tried to develop an efficient process that could enable fluoropyridine hydrogenation by overcoming catalyst poisoning and loss of the fluorine atoms. Keeping this in mind, we thought that adding an initial step that could first break the aromaticity of the substrates, forming a mixture of diene intermediates, would facilitate the hydrogenation reaction.
Motivated by this idea, we started looking for a reaction model to prove our theory. Some literature reports on rhodium-catalyzed pyridine dearomatization suggested pinacol borane (HBpin) as the reagent of choice. We set up a reaction vessel containing 3-fluoropyridine, HBpin, THF, molecular sieves and a rhodium-carbene catalyst. We added molecular hydrogen and heated the mixture to room temperature. As it happens, that worked out very well! We further optimized the reaction conditions for the dearomatization-hydrogenation (DAH) process and admittedly, that took a while! All reagent and solvents must be dry prior the reaction, otherwise formation of the hydrolyzed species of HBpin can deactivate the catalytic cycle.
Having everything optimized, we were able to access the all-cis-fluorinated piperidine derivatives in a highly diastereoselective fashion. Bear in mind that the newly developed protocol consists of a one-pot two-step reaction, where as previously, synthesis of (multi)fluorinated piperidines required tedious multistep protocols.
Our method allows the formation of highly desirable products that are of great interest for medicinal, agrochemistry and materials science. But fundamentally, even more interesting was the study of the conformational behavior of fluoropiperidines in solution by NMR spectroscopy. A first, comprehensive study determining the axial/equatorial orientation of fluorine-substituents was enabled.
Are you curious about more details on the DAH process? You can find the full paper here.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in