

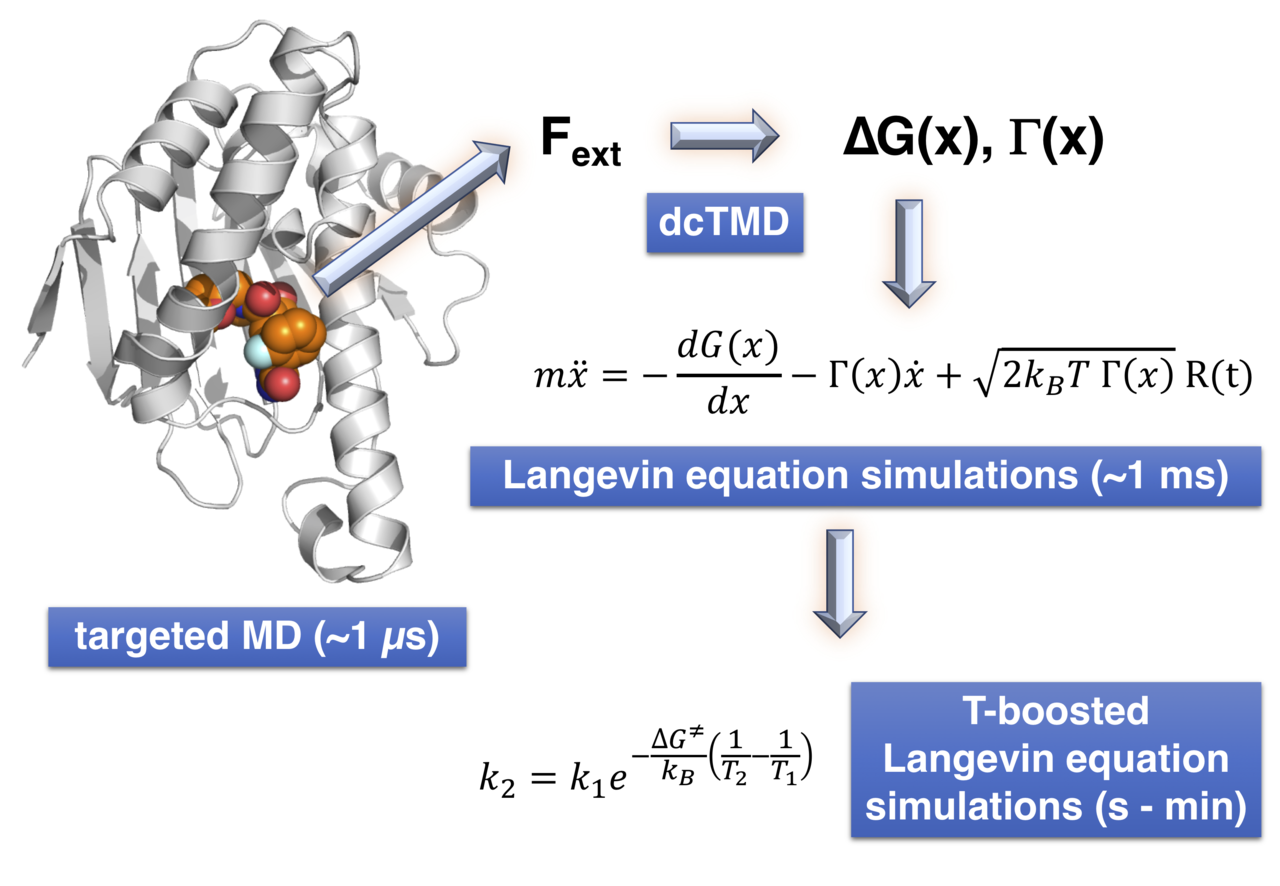
Fig. 1: workflow of dissipation-corrected targeted MD in combination with temperature-boosted Langevin equation simulations.
Molecular Dynamics (MD) simulations have become ubiquitous in life science for the last decades. The simulation community currently faces two major challenges: accessing biologically relevant length and timescales.
Length scales are rather straightforward to access by using computers with increasing processing capacity: the largest system simulated so far is a 1 billion atom representation of an entire gene locus (GATA4)1. Progress on longer time scales is facing significant problems: as MD simulations require an iterative integration of Newton’s equations of motion, only the creation of a faster processors can achieve longer calculations, but CPU frequencies have been stuck at a limit of 4 GHz for more than a decade now. While the construction of dedicated hardware has allowed to access the milliseconds regime2, processes that need seconds and beyond such as protein-drug binding and unbinding are still far out of reach.
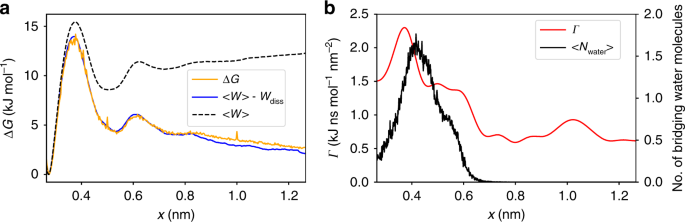
A viable approach to accelerate simulations is to coarse-grain the full system dynamics, which is a domain of nonequilibrium statistical mechanics. A central requirement is the presence of a timescale separation between slow (e.g., protein-ligand unbinding) and fast processes (e.g., protein vibrations and water fluctuations): then, the Langevin equation can be used to express the dynamics of the system along a reaction coordinate such as the distance of the ligand from its binding site. Slow degrees of freedom are contracted into a free energy, and all fast degrees of freedom into a Stokes-type friction coefficient together with a random fluctuating force. To perform the necessary coarse-graining, we recently developed dissipation-corrected targeted MD (dcTMD)3: applying a constraint force to actively pull a system along a coordinate of interest4, the necessary pulling work can be decomposed into free energy and fiction fields of the process.
In our new work represented here5, we now show these dcTMD fields can be used as input for a Langevin equation simulation along the pulling coordinator. The advantage of this approach is a drastic reduction in necessary computing power: 1 ms of simulated time can be reached within several hours on a single core of a standard desktop computer. A second feature of the Langevin fields allows for additional acceleration: unlike fully atomistic proteins, fields do not denature at higher temperatures. High temperature simulations therefore can produce “boosted” dynamics that can be used to recover dynamics at a lower temperature of interest under which the fields were derived from targeted MD simulations5.
Using the dissociation of NaCl and of two protein-ligand complexes as test systems, we show that we can predict dynamics of binding and unbinding processes on a timescale of seconds to half a minute. While the Langevin fields were generated only from unbinding simulations, we can predict both unbinding and binding kinetics within a factor of 20, and dissociation constants within a factor of four, which is within the best range achievable6. At the same time, our dcTMD approach requires up to a magnitude less computational power than other coarse-graining methods7.
Last but not least, the determination of friction profiles allows for gaining insight into molecular processes that the free energy is oblivious to: we found that in all investigated systems, the formation of a water hydration sphere appears to form the main source of friction, which allows for the derivation of new principles for the design of drugs with desired binding and unbinding kinetics.
1. Jung, J. et al. Scaling molecular dynamics beyond 100,000 processor cores for large‐scale biophysical simulations. J. Comput. Chem. 40, 1919–1930 (2019).
2. Lindorff Larsen, K., Piana, S., Dror, R. O. & Shaw, D. E. How Fast-Folding Proteins Fold. Science 334,517–520 (2011).
3. Wolf, S. & Stock, G. Targeted Molecular Dynamics Calculations of Free Energy Profiles Using a Nonequilibrium Friction Correction. J. Chem. Theory Comput. 14, 6175–6182 (2018).
4. Schlitter, J., Engels, M. & Krüger, P. Targeted molecular dynamics: a new approach for searching pathways of conformational transitions. J. Mol. Graph. 12, 84–89 (1994).
5. Wolf, S., Lickert, B., Bray, S. & Stock, G. Multisecond ligand dissociation dynamics from atomistic simulations. Nat. Commun. 11, 2918 (2020).
6. Bruce, N. J., Ganotra, G. K., Kokh, D. B., Sadiq, S. K. & Wade, R. C. New approaches for computing ligand-receptor binding kinetics. Curr. Opin. Struct. Biol. 49, 1–10 (2018).
7. Nunes-Alves, A., Kokh, D. B. & Wade, R. C. Recent progress in molecular simulation methods for drug binding kinetics. arXiv.org, arXiv:2002.08983 (2020).



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in