
It is well known that, for the industrial heterogeneous ammonia synthesis catalyzed by fused Fe or supported Ru catalysts, N2 undergoes dissociative activation into surface N atoms followed by stepwise hydrogenation to NH3.1,2 The high kinetic barrier for direct N2 dissociation and/or strong adsorption of NHx species on Ru or Fe metal surfaces render the formation of ammonia under forcing conditions (> 673 K, > 100 bar).3 While for the homogenous process catalyzed by well-defined molecular TM complexes, the reduction of N2 to NH3 occurs under ambient or cryogenic conditions by using excess proton sources and reducing reagents, and is believed to proceed via the hydrogen-associative mechanism.4 Nevertheless, it requires large energy inputs as a consequence of the competing hydrogen evolution reaction. Inspired by the knowledge and insights from these two manmade landmark systems, one may wonder whether it is possible to achieve non-dissociative hydrogenolysis of N2 to NH3 via concerted N-N weakening and N-H bond formation, thereby minimizing the energy cost to facilitate ammonia production under mild conditions. Despite some encouraging attempts have been made in both heterogeneous and homogeneous regimes,5-7 the catalytic hydrogenolysis of N2 to NH3 by using H2 as the reductant is one of the unresolved challenges in chemistry. Breakthroughs in catalyst development are highly needed!
Our journey towards complex hydrides for N2 activation and ammonia synthesis started in year 2016. Our previous studies showed that composite catalysts comprising LiH and 3d metals (Fe, Co, Mn, etc.) are highly active for ammonia synthesis,8 and gas-phase cluster investigations revealed that laser-generated gaseous clusters such as [Li4FeH6]ˉ and [Li5FeH7]ˉ from the LiH-Fe composite catalyst can undergo stoichiometric reaction with N2 to form NH2-containing clusters.9 These findings stimulated us to hypothesize that complex hydrides of alkali (alkaline earth) metals and transition metals may be effective in catalyzing ammonia synthesis. Unfortunately, we cannot synthesize solid-state Li4FeH6 in our lab due to the need of extremely high hydrogen pressure (ca. 60000 bar).10 Through the literature research, we found that its Ru analogues (such as Li4RuH6 and Ba2RuH6) can be facilely synthesized by reacting Ru metal and LiH or BaH2 under normal conditions, and are a class of well-characterized complex hydrides.11
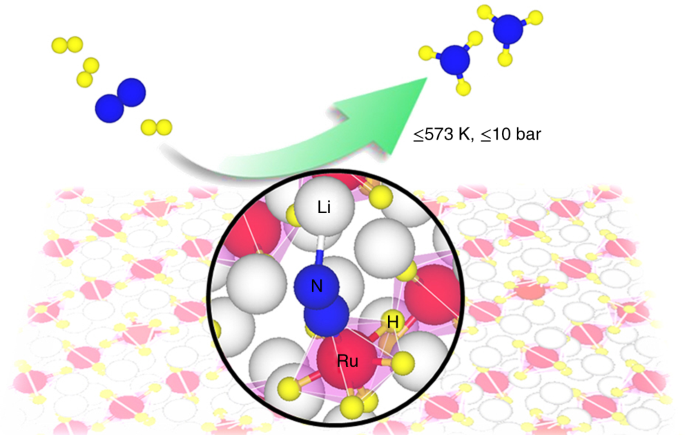
For Li4RuH6 or Ba2RuH6, Ru is in ionic state and coordinated with multiple hydridic hydrogens, creating an electron-rich environment for N2 reduction. Besides, the Li or Ba cations form a K4CdCl6 (or K2PtCl6)-type framework to spatially isolate the electron-rich [RuH6]4ˉ anions, resulting in a Ru-Ru distance higher than 5.00 Å that is unfavorable for dissociative N2 adsorption (Figure 1a). Such a unique configuration differs distinctly from that of closely packed Ru metal surface (Figure 1b) which has a short Ru-Ru distance of 2.71 Å and thus can utilize several adjacent Ru atoms (such as B5 site) to induce the cleavage of N2, but share some similarities with that of the molecular Ru complex catalyst (Figure 1c) which has isolated and ionic Ru centers for associative N2 activation. Such characteristics of the solid-state Ru complex hydrides aroused our great interest to investigate their performance in catalyzing N2+H2 to NH3.
To our delight, continuous NH3 formation was observed by directly applying Li4RuH6 or Ba2RuH6 as the catalyst. Notably, the operating temperatures of MgO-supported Li4RuH6 (Li4RuH6/MgO) or Ba2RuH6 (Ba2RuH6/MgO) are substantially downshifted by ~100 K from the benchmark Ru/MgO catalyst. At 573 K and 1 bar, Li4RuH6/MgO and Ba2RuH6/MgO outperform the Ru/MgO by 24 and 36 times, respectively (Figure 1d). It is worth noting that Ba2RuH6/MgO is capable of catalyzing N2+H2 to NH3 at temperatures below 373 K (Figure 1e). Despite at a low-level rate, this is an encouraging step toward near-ambient ammonia synthesis.

The unique configuration and superior catalytic performance of the complex hydride catalyst motivated us to explore its function mechanism for catalyzing ammonia synthesis. Given the complex nature of the ternary hydride surface, DFT calculations are very helpful in identifying the atomic-level mechanistic details. Taking the Li4RuH6 catalyst as an example, we collaborated with Prof. Tejs Vegge, Dr. Heine Anton Hanson, and Dr. Jaysree Pan from Technical University of Denmark to elucidate its microscopic reaction mechanism with a combined experimental and computational approach. In the past three years, we held meetings every two weeks to discuss the progress from theory and experiment, and investigated a large number of possible pathways to pursue an optimum one. Finally, a kinetically balanced non-dissociative path that involves the formation of N2Hy (y = 1-3) species followed by N-N bond breaking was demonstrated to be more energetically favorable. Such a path is enabled by the synergistic engagement of all elements of the complex hydride: the electron-rich [RuH6]4ˉ anionic center mediates N2 reduction, the lattice hydridic hydrogens are proton and electron carriers, and the Li cations stabilize the NxHy (x = 1-2, y = 1-3) intermediate species. In parallel, some important mechanistic features (such as the non-dissociative N2 activation and the involvement of lattice hydridic hydrogens) were supported by designed experiments, and several reacting species (such as the chemisorbed N2 and NH2 species) along the pathway was trapped by experimental characterization. For instance, the 15N2-14N2 isotopic exchange reaction (IER) experiment shows that the Li4RuH6/MgO catalyst has negligible IER rates, illustrating a non-dissociative pathway prevails during catalysis.
Based on the systematic and detailed research depicted above, we realized the key to achieving efficient NH3 synthesis lies in the unique configuration and function mechanism of ruthenium complex hydride, which differs fundamentally from the conventional and recently-developed heterogenous transition metal catalysts. The solid-state Ru complex hydride described here serves as the very first case that realizes catalytic hydrogenolysis of N2 to NH3 by using H2, which sets up a bridge linking research endeavor from homogeneous and heterogeneous nitrogen fixation.
With the beauty, complexity, and potency of the catalysis disclosed, this study demonstrates the power of multi-component and electron-rich catalytic center to tackle the activation and conversion of kinetically inert N2. It also implies that the complex hydride may serve as a unique heterogeneous catalyst for other important chemical transformations that require electron and proton inputs.
More details can be found through the link: https://doi.org/10.1038/s41929-021-00698-8
References
1 Jennings, J. R. Catalytic Ammonia Synthesis: Fundamentals and Practice 1st edn (Springer, 1991)
2 Honkala, K. et al. Ammonia Synthesis from First-Principles Calculations. Science 307, 555-558, doi:10.1126/science.1106435 (2005).
3 Vojvodic, A. et al. Exploring the limits: A low-pressure, low-temperature Haber-Bosch process. Chem. Phys. Lett. 598, 108-112, doi:10.1016/j.cplett.2014.03.003 (2014).
4 Chalkley, M. J., Drover, M. W. & Peters, J. C. Catalytic N2-to-NH3 (or -N2H4) Conversion by Well-Defined Molecular Coordination Complexes. Chem. Rev. 120, 5582-5636, doi:10.1021/acs.chemrev.9b00638 (2020).
5 Pool, J. A., Lobkovsky, E. & Chirik, P. J. Hydrogenation and Cleavage of Dinitrogen to Ammonia with a Zirconium Complex. Nature 427, 527-530, doi:10.1038/nature02274 (2004).
6 Avenier, P. et al. Dinitrogen Dissociation on an Isolated Surface Tantalum Atom. Science 317, 1056-1060, doi:10.1126/science.1143078 (2007).
7 Shima, T. et al. Dinitrogen Cleavage and Hydrogenation by a Trinuclear Titanium Polyhydride Complex. Science 340, 1549-1552, doi:10.1126/science.1238663 (2013).
8 Wang, P. et al. Breaking Scaling Relations to Achieve Low-temperature Ammonia Synthesis through LiH-mediated Nitrogen Transfer and Hydrogenation. Nat. Chem. 9, 64-70, doi:10.1038/nchem.2595 (2017).
9 Wang, P. et al. The Formation of Surface Lithium-Iron Ternary Hydride and its Function on Catalytic Ammonia Synthesis at Low Temperatures. Angew. Chem. Int. Ed. 56, 8716-8720, doi:10.1002/anie.201703695 (2017).
10 Saitoh, H. et al. Li4FeH6: Iron-containing Complex Hydride with High Gravimetric Hydrogen Density. APL Mater. 2, 076103, doi:10.1063/1.4886219 (2014).
11 Humphries, T. D., Sheppard, D. A. & Buckley, C. E. Recent Advances in the 18-electron Complex Transition Metal Hydrides of Ni, Fe, Co and Ru. Coord. Chem. Rev. 342, 19-33, doi:10.1016/j.ccr.2017.04.001 (2017).




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in