Chemotherapy is highly efficient and widely used for cancer treatment. However, many commercially applied chemotherapeutic drugs lack the selectively between cancer and normal tissues, and thus rise systemic toxic effects to patients. Prodrug is known to be an effective strategy to reduce this side effect and enhance selectivity. Typically, prodrugs are chemically modified drug molecules which release pharmacologically active drugs upon transformation in vivo. Benefiting from advanced organic synthesis and biosynthesis techniques, active sites of pharmaceutically active drugs can be modified to achieve non-toxic prodrugs and therefore shield their toxicities to minimize the required dosage and reduce adverse side effects to patients. When the prodrugs reach tumor areas, stimuli triggered deshielding of the modification groups take place and release the activated drugs, thus achieved a precise tumor treatment.
The stimuli used to activate prodrugs can be divided into two categories, namely internal stimuli and external stimuli. The internal stimuli mainly include over-expressed enzymes, redox species, low pH, and hypoxia in the tumor microenvironment. External stimuli often offer precise temporal and spatial control over site of action. For example, light, ultrasound, heat, and synthetic molecular injections can trigger prodrug activations in high efficiency at the site of disease and at a desired period of time.
Radiotherapy is an effective treatment strategy for inhibiting tumor growth and is often used in combination with chemotherapy to treat patient suffering from advanced cancers. Concomitant chemo-radiotherapy for malignant tumors originates from the concept of comprehensive treatment and has become a standard treatment method to prolong patients’ survival time. Most currently used concomitant treatments applies sensitizer-like chemical drugs to promote the effect of radiotherapy. However, the use of radiotherapy radiation sources, i.e. X-rays, as a source of stimuli to activate prodrugs, is still rarely investigated.
We can imagine, if an anti-tumor drug can be converted into an X-rays activatable prodrug with sufficient stability and significantly reduced toxicity, the systemic toxicity issue can be overcome. For patients received a dosage of such prodrug, toxic drug molecules will only be generated at the tumour site when receiving the X-ray treatment. Additionally, with the development of therapeutic X-ray techniques, precise radiation with high resolution can be achieved, so that the prodrug activation region may be precisely controlled. Therefore higher doses of prodrugs may be applied for patients to further improve the efficacy.
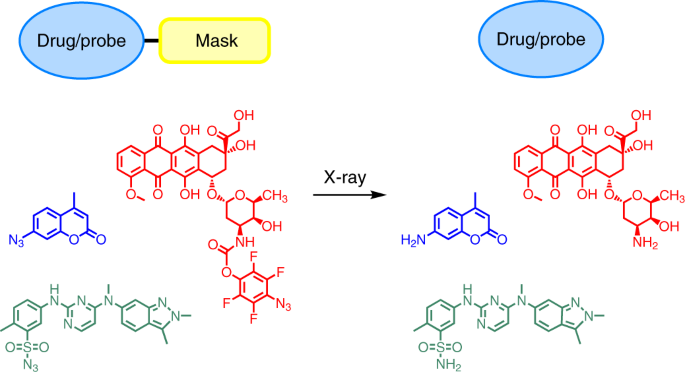
Here, we have explored the possibility of using clinically utilized radiotherapy X-ray sources to activate anti-tumor prodrugs. In the early stage of this project, we have conducted a wide-range screening over a series of potentially activatable organic molecules that may react upon X-ray radiations. Fortunately, we found sulfonyl azides and fluoroaryl azides can be converted to corresponding sulfonamides and aromatic amines at clinically relevant doses of X-ray radiation in high efficiency. It is reported that X-ray radiation of aqueous solutions can generate reactive species such as hydroxy radicals, hydrogen radicals, hydrogen peroxide, etc. In combination with our experimental findings, we propose that the azide reduction mechanism was through a sulfonylamido radical or a nitrene intermediate.

Figure 1. A coumarin-based quenched fluorophore and pazopanib and doxorubicin derived model prodrug molecules were activated by X-ray radiation.
To translate these findings to medical applications, we introduced the functional groups to antitumor drug molecules and fluorescent reporters to synthesize a series of prodrugs and quenched fluorophores. We verified their activation efficiency, pharmacological activity and biosafety in vitro and in vivo. We found the prodrug molecules can efficiently reduce systemic toxicity while maintaining the efficacy of the parent drugs, and thus significantly prolong the survival period of the tumour bearing animals. We believe this X-ray activatable prodrug system could represent an important modality within therapeutic oncology and open up a new era in targeted and directed cancer chemoradiotherapy.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in