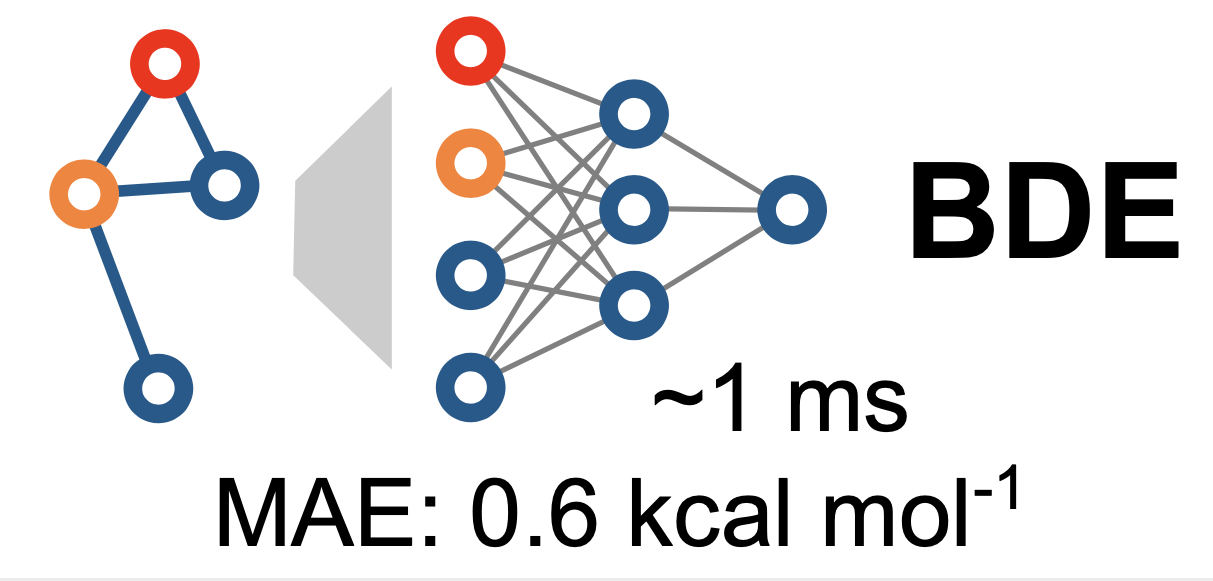


For this paper, a collaboration between Peter St. John, Seonah Kim and Yeonjoon Kim at NREL and Yanfei Guan and Robert Paton at Colorado State University, we trained a graph convolutional neural network against nearly 300,000 organic bond dissociation enthalpies. The resulting model, a machine-Learning derived, Fast, Accurate Bond dissociation Enthalpy Tool (ALFABET) predicts BDE values that are comparable to density functional theory in much less than a second. The predictions can be run on the web: https://bde.ml.nrel.gov/

Machine learning approaches such as ours are “data hungry” so we needed a large number of BDE values to train the model. Far more than are available experimentally, in fact. We compared a variety of quantum mechanical methods against the very useful iBonD database and found that M06-2X/def2-TZVP calculations agreed most closely. Using this level of theory we built an automated workflow to fragment around 40,000 organic molecules in every way conceivable* and to launch and analyze hundreds of thousands of DFT calculations required to obtain BDE values. Fragmentation and conformational analysis were done using Python and rdkit. In the paper we describe the effects of using multiple DFT conformers vs just one on the results. Running this many calculations is challenging, particularly when they involve open-shell species (e.g., radicals) , since there are various errors that must be checked and filtered in an automated fashion. In addition, we used a statistical model to detect for the presence of outliers in the computational data that were then removed.
To exemplify how ALFABET can be used, we predict the weakest C-H bond(s) in a series of pharmaceutical molecules, and show that these sites are highly represented among the positions of metabolism (e.g., through oxidation P450 enzymes). The analysis takes seconds and gives comparable results to much lengthier DFT calculations. We also show that for a series of molecules used in fuel, the identities of radicals formed by cleavage of the weakest bond can be used to develop a multivariate linear regression model that predicts the yield sooting index, a measurement of soot formation. Future extension of the ALFABET tool with additional DFT calculations will enable predictions for expanded atom types and zwitterionic functional groups.

Although the training data was generated using 3D structures, the graph neural network employed in this study only requires the 2D-connectivity of each molecule, which performs adequately for this task. Other properties, such as NMR chemical shift, require the explicit consideration of different molecular conformers, and therefore require 3D atomic features.. We have begun to explore this area.
* With the exception of double bonds, and rings, and fragmentations that create new stereogenic centers (e.g. through the destruction of symmetry)
Read the paper: Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost Peter C. St. John, Yanfei Guan, Yeonjoon Kim, Seonah Kim & Robert S. Paton, Nat. Commun. 2020, 11, 2328




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in