
OLED general remarks Organic light emitting diodes (OLEDs) have been recognized as a fast-emerging technology with an immense application potential for displays of superior quality. In fact, OLEDs are currently operated in commercialized consumer electronics, including in high-end iPhone and Android device displays, where they’re known for their dark blacks and high-visibility in sunlight. Apart from displays, further applications are being explored and entail, for instance large area lighting or smart labels for innovative packaging. The possibility of the deposition of electroluminescent substances on extraordinary thin layers gives rise to unprecedented flexibility in display- and panel-lighting designs, even if the current generation of folding-phone displays haven proven to be a bit unreliable! In this respect, a great deal of attention is payed to luminophores based on heavy expensive transition metal complexes (Ir, Pt, Ru) as they allow for very efficient luminescence due to strong emission of their lowest excited triplet state. They are often referred as triplet-harvesters and, when used in an OLED, they are also known as PHOLEDs (phosphorescent OLEDs). Clearly, it is of high interest to substitute expensive scarce elements for more cost-efficient abundant alternatives, especially with respect to additional future mass-market applications1 Against this background, copper(I) coordination compounds are the subject of recent investigations both in academia and industry. Copper is 3 to 5 orders of magnitude more abundant in the Earth’s crust than some of the heavier elements that are often used in OLEDs, making it not only more accessible but much cheaper. Heavier transition metal complexes showing strong spin-orbit interactions which give rise to efficient triplet harvesting due to an intersystem crossing from the excited singlet to the lower lying triplet state (phosphorescence). This triplet state phosphorescence is long-lived, making it attractive for display technologies. In contrast, the remarkable photo luminescence of many copper(I) compounds stems from the so-called thermally activated delayed fluorescence effect (TADF).2 That is, a suitable small singlet-triplet gap allows for a thermally activated back-transition from the triplet to singlet state (reverse intersystem crossing, rISC) and thus efficient emission via prompt- and delayed fluorescence.
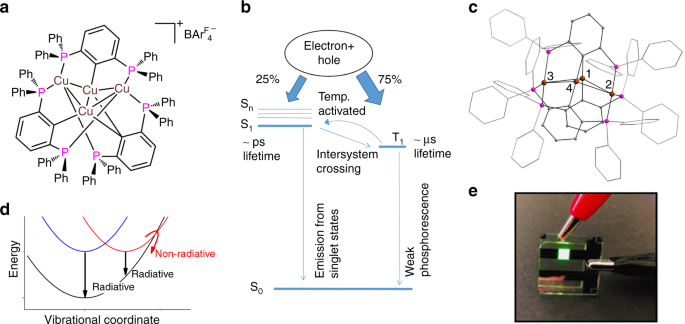
Synthesis of [Cu4PCP)3]+ While carbo-metallated complexes of precious metals are widely used as luminophores in electroluminescent devices, reports of organometallic copper complexes for such applications are very rare. This may be due to synthetic difficulties; especially when processed in solution, such compounds show often dynamic coordinative behavior. The air and moisture sensitive Cu–C bond can impose additional handling problems. We have recently reported the synthesis of a small cationic Cu4 cluster [Cu4(PCP)3][A] with PCP= 2,6,-(PPh2)2C6H3, A= BF4– or BArF4–) as a rare example of an cationic organometallic copper complex showing intense green emission in the solid as well as solution (Figure 1, A and B, respectively). The cluster complex can serve as robust luminophore in solution processed OLED prototype devices.3 Specifically, the reaction of [Cu(CH3CN)4]BF4 and the ligand precursor 2,6,-(PPh2)2C6H3SiMe3 in a 4:3 ratio allows for C–Si bond cleavage and C–Cu bond formation under concomitant release of FSiMe3 while BF4– serves as the mild source of F–. As a result, the tetranuclear cationic cluster [Cu4(PCP)3][BF4] is formed (Figure 1, D). The crystal structure shows a rigid structure having a distorted kite-type Cu4 core with one of the shortest Cu–Cu interatomic distances, which is chelated by three anionic PCP tridentate ligands. As a result, the two central Cu centers are solely coordinated by carbon (phenylate, C-domain) capped by two phosphine-only (P-domain) coordinated copper centers. The resulting overall cationic species is balanced by a BF4– counter anion. (Figure 1, E). Anion exchange to BArF gives rise to a highly soluble compound. The route avoids common problems of standard transmetallation reactions, such as contamination with halides and aggregation with other metal salts, as anionic cuprates in solution show often formation of contact- or solvent ion pairs.
Utilization of [C4PCP)3]+ in prototype OLED [Cu4(PCP)3]+ is remarkably robust: dynamic ligand exchange or dissociation with associated solvent coordination was not detected in a broad range of solvents. [Cu4(PCP)3]+ exhibits PL efficiency of up to 93% (in frozen solution) and narrow emission with a full width at half maximum (FWHM) of 58 nm (solid) and 60 nm (solution). Prototype OLED devices were prepared exploiting [Cu4(PCP)3]+ as solution-processed luminophore for the emissive layer (Figure 1, F). Typical devices built exhibit a very narrow green emission (65 nm FWHM) with resultant CIE 1931 chromaticity coordinates of [0.305, 0.637] at 1000 cd m-2 exceeding the primary green sRGB (BT.709) color standard.

Figure 1: A solid [Cu4(PCP)3]+ , B [CU4(PCP)3]+ in THF, C Jet set-up within experimentation chamber at SWISS FEL, D synthesis scheme of [Cu4(PCP)3]+ , E crystal structure of [Cu4(PCP)3]+ , F solution processed OLED prototype device.
As you can see in Figure 1 E, the structure of [Cu4(PCP)3]+ is an important aspect of its unique performance. What’s interesting about this particular material is how the thermally-activated delayed fluorescence works and whether we can obtain information on the dynamic structure of the molecule in operando so to speak. To do this there are a range of techniques available, including theoretical simulation approaches, but the ones we chose to use, took advantage of pulsed X-ray sources to use short wavelength photons to probe the molecule in solution. X-rays are particularly powerful since they can provide atomic-scale information on the sample, and because they’re quite penetrating, they can look at systems in solution easily. The two techniques used in these measurements are based on X-ray scattering and X-ray spectroscopy. X-ray scattering measures the X-rays elastically scattered from the sample, which is directly related to the position of all the atoms in the scattering volume. X-ray spectroscopy uses tunable, monochromatic X-rays to probe specific elements in the sample. By measuring the X-ray absorption spectrum of a sample you can obtain information on both the structure and electronic environment around the absorbing atom. A complementary technique to X-ray absorption is X-ray emission, where a spectrometer is used to measure the X-ray fluorescence spectrum from the sample. All of these methods can be used in a so-called pump-probe fashion, where you use an optical laser to excite the sample and then you probe it using an X-ray pulse. Below you can find a short movie clip illustrating this technique: You can see a magnified image of the running jet-system in the experimental chamber of SwissFEL user facility (see below), where [Cu4(PCP)3]+ is dissolved in an organic solvent and circulated using an HPLC pump. When the optical laser hits the jet, the solution brightly illuminates. This is visible as a flashing green light (Movie Clip 1).
Movie Clip 1. Swiss FEL jet-installation and its interaction with the optical laser.
X-ray facilities The two types of large-scale X-ray user facilities that exist around the world for researchers to use, are storage ring synchrotron light sources and X-ray free electron lasers (XFELs). Storage rings produce light ranging from IR all the way through to hard X-rays, and though most experiments take advantage of the average X-ray photon flux at a synchrotron beamline, the actual time structure of the X-ray beam is pulsed, often consisting of a flash of X-rays about 100 ps long and repeated every 2 ns or so. XFELs are a newer and quite different method of producing short wavelength light, which results in very short bursts of X-rays, often around 50 fs long. Most current XFEL facilities work at lower repetition rates around 100 Hz, though a few projects can or will operate at higher repetition rates. In general, this means that for time-resolved experiments you can measure dynamics from 100 ps out to the microsecond or millisecond regime at storage rings, and XFELs can provide information down to a few tens of fs. For these experiments we went to three different X-ray facilities: the ESRF synchrotron light source (Grenoble, France), the SLSsynchrotron light source (Villigen, Switzerland), and the SwissFEL X-ray free electron laser, (Villigen, Switzerland). At the ESRF we measured X-ray scattering from a flowing solution of [Cu4(PCP)3]+ on timescales ranging from 100 ps through to 2 μs after the pump laser pulse4. This allowed us to look at the structure of the molecule in the TADF triplet state, but couldn’t give us any information on the short-lived singlet state, which is actually responsible for the emission. At the SLS we measured the X-ray absorption spectrum5 around the Cu K-edge at 8.98 keV, where the X-rays are resonant with the Cu 1s electron. This measurement was also performed 1 μs after photoexcitation of the OLED luminophore and gave information both on the TADF triplet state structure, but also on the changes in electronic density around the Cu atoms in [Cu4(PCP)3]+. The last experiment took place at the new XFEL facility, SwissFEL6, and took advantage of its ability to cover the so-called tender X-ray regime (2-5 keV), which covers a range of the periodic table including P, S, Ca and the 4d transition metals (e.g. Ru, Rh, Pd, Ag). At SwissFEL we used a technique called non-resonant X-ray emission spectroscopy, where the incident X-rays are kept at a fixed energy and the X-ray emission spectrum is measured using a single-shot dispersive X-ray spectrometer7. For these experiments we were looking at X-ray emission from the P moiety of [Cu4(PCP)3]+ , and how the electronic environment of the P atoms are changed when the luminophore is excited into the triplet state. By combining all these different techniques (measured X-ray signals are exemplary shown in Figure 2) we were able to get a picture of how [Cu4(PCP)3]+ functions in the triplet excited state from a viewpoint concerning the geometrical - and electronical structural change to better be able to understand how the TADF process affects the luminescent properties of the molecule.

Figure 2: The X-ray signals measured in these experiments. From left to right they are: The phosphorus non-resonant X-ray emission, where the top figure shows the ground-state Kα1,2 X-ray emission (black curve) and the bottom shows the difference between the laser excited and ground-state spectrum (black markers), overlaid with the simulated difference spectrum expected in the triplet state (blue curve). The middle figure shows the copper K-edge X-ray absorption near-edge structure (XANES) spectrum, with the top (black) showing the pump probe difference spectrum measured 1 μs after photoexcitation, the middle (blue) curve shows the ground-state XANES spectrum, and the bottom shows the simulated ground-state XANES spectra for the two coordination states of Cu in [Cu4(PCP)3]+ , one of which is coordinated to C and the other to P atoms. The right figure shows the X-ray scattering difference signals at various times delays after excitation along with the simulated scattering difference signal from DFT calculations (red curve) and the simulated difference signal taking into account the effect of solvent heating (blue curve).
Taking Snapshots For single-core copper-based luminophores the mechanism of emission is usually a result of a so-called metal to ligand charge transfer (MLCT) process. That is, upon absorption of light, charge transfer is observed from the metal center to a suitable ligand centred empty orbital. However, [Cu4(PCP)3]+ is a multi-core complex and therefore significantly different. In particular, 2x2 copper centers reside in different chemical environments: the C- and P-domain, respectively, having metal-metal interactions in between each other. This special structure of [Cu4(PCP)3]+ presented three aspects of interest to us, which we could tackle with the powerful combination of the pump-probe X-ray scattering and spectroscopy techniques:
- Which exact Cu centers participate in the MLCT process? C-coordinated, P-coordinated or all together?
- What about the coordinating P atoms? Do they contribute additionally to the charge transfer process or does their role remain predominantly to the strong Cu-coordination in the P-domain, providing a rigid and robust structure?
- How is the structure of the Cu4-core changing as a result of the excitation?
To sum up, our experiments suggest that MLCT occurs rather delocalised over the whole Cu4-core and interestingly, the P-atoms are involved in the charge shift as well. We see marginal changes of the Cu-Cu interatomic distances in the triplet state. This means that the rigid cluster motif prevents major distortion in the excited state, which prevents non-radiative losses. This is an important factor for an efficient emission. In that sense, the multicore-approach in [Cu4(PCP)3]+ may complement other approaches in copper(I) emitters, such as steric hinderance, or reduction in coordination number, to maximise structural rigidity and lowering the reorganization energy.
If you want to read more about our project you can now find our publication "Taking a snapshot of the triplet excited state of an OLED organometallic luminophore using X-rays" online at Nature Communications 11, 2131 (2020) doi.org/10.1038/s41467-020-15998-z
Christopher Milne & Matthias Vogt
1. Bizzarri, C. et al. Sustainable metal complexes for organic light-emitting diodes (OLEDs). Coordination Chemistry Reviews (2018). https://doi.org/10.1016/j.ccr.2017.09.011
2. Czerwieniec, R. et al. Cu(I) complexes – Thermally activated delayed fluorescence. Photophysical approach and material design. Coord. Chem. Rev. 325, 2–28 (2016). https://doi.org/10.1016/j.ccr.2016.06.016
3. Olaru, M. et al. A Small Cationic Organo-Copper Cluster as Thermally Robust Highly Photo- And Electroluminescent Material. J. Am. Chem. Soc. 142 373-381(2020) https://doi.org/10.1021/jacs.9b10829
4. Haldrup, K., Christensen, M., Nielsen, M. Analysis of time-resolved X-ray scattering data from solution-state systems Acta Crystallographica Section A 66(), 261 - 269 (2010). https://dx.doi.org/10.1107/s0108767309054233
5. Smolentsev, G. et al. X-ray absorption spectroscopy with time-tagged photon counting: Application to study the structure of a Co(I) intermediate of H2 evolving photo-catalyst Faraday Discussions 171(0), 259 - 273 (2014). https://dx.doi.org/10.1039/c4fd00035h
6. Milne, C. et al. SwissFEL: The Swiss X-ray Free Electron Laser Applied Sciences 7(7), 720 (2017). https://dx.doi.org/10.3390/app7070720
7. Milne, C. et al. Opportunities for Chemistry at the SwissFEL X-ray Free Electron Laser CHIMIA International Journal for Chemistry 71(5), 299-307 (2017). https://dx.doi.org/10.2533/chimia.2017.299




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in