
Virtual Chemist is an idea that took root in my mind some 20 years ago. It’s an idea that has grown slowly ever since, with the pieces of the puzzle coming together over time. The story of Virtual Chemist is one of perseverance and progress made in a field of research that was slow to accept such an idea. It’s a story of a university that had faith in me and helped me bring the idea to fruition.
The story began during my PhD at the University of Nancy, France (1995-1998). The main objective of my doctorate was to develop analogues of a natural product, adenophostin A. As part of my efforts to synthesize these substances, I employed the well-known Sharpless asymmetric dihydroxylation (AD) reaction. The use of AD led to an uncommon mismatched case, in which dihydroxylation of a xylose derivative led to the stereoisomer opposite to that predicted by the Sharpless mnemonic (1996-1997). Working at the interface of organic and computational chemistry under the supervision of Dr. Yves Chapleur (carbohydrate chemistry) and Dr. Bernard Maigret (computational chemistry and biochemistry), I thought that computational chemistry might offer some insight into this surprising result. The dual training I was receiving was key to an important realization on my part: developing an idea of this kind would only be possible with the right support and a background in both disciplines. Thanks to my supervisors, I made the first steps towards developing the first component of the platform that would eventually become Virtual Chemist.
While I contemplated QM/MM, the use of pure molecular mechanics (MM) was an attractive alternative which would enable a more efficient conformational sampling. The question was really about modeling transition states (TS) with MM. In 1999, Per-Ola Norrby’s et al. publication came to my aid in what turned out to be a second turning point in my career. Where Houk had proposed modelling TS as rigid templates in the early 1980s, Norrby presented what came to be one of the most influential papers on TS modeling using MM. His methods applied specifically to Sharpless dihydroxylation (J. Am. Chem. Soc. 1999, 121, 10186-10192)! My mind was made up: I would model TS using MM!
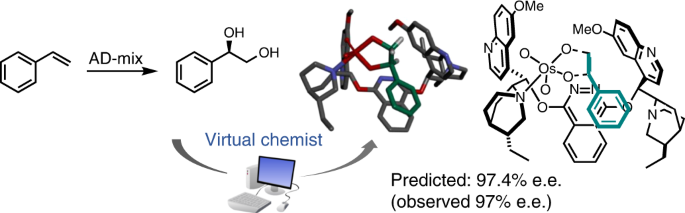
With some experience in conformational searches, my first thought was to develop a genetic algorithm-based approach to optimize TS structures of the dihydroxylation of olefins using large catalysts such as the one included in the widely used AD-mixes. In this first version (2002), the TS was kept frozen. While I initially thought this approach would provide good quality TS structures (qualitative), it turned out – unexpectedly – that the accuracy in predicting the stereochemical outcome was actually quite good.
At the time, I did not fully appreciate the potential of such an approach, thinking I had simply found a good explanation for an unusual reaction behavior. That was until I was contacted by a colleague in France. While he found our mismatched case intriguing, he was even more interested in our method: could I apply it to his carbohydrate derivatives which abided by the mnemonic? This was an epiphany: the method went beyond dihydroxylation and had great potential for asymmetric catalyst design. This coincided with my move to McGill University (Montreal) and the concept was at the core of one of my research proposals, which could be summed up as: Computer-aided drug design, I can do it; computer-assisted catalyst design, I will do it! And McGill believed me.
Surprisingly, while computer-aided medicinal chemistry was well funded in my lab, the concept of computer-assisted catalyst design was not well regarded by the research community. I soon realized that while the pharma industry had been quick to adopt the use of computers in medicinal chemistry, the major developments in asymmetric synthesis had mostly been achieved by academic groups. Several discussions with experienced organic chemists confirmed my impression. The apprehension that computers would replace, not assist, chemists was strong and it was accompanied by the belief that chemists could be creative without the need for computers.
I saw things differently, believing that, in a computational environment, chemists would be able to test their ideas faster and therefore be a lot more creative. But it was a few years before I was able to secure a small grant to recruit a student to help me in these efforts. At the time, we had a method for performing conformational searches; what we needed was a way to render TSs flexible. As an organic chemist, I see a TS as something somewhere between reactants and products. It sounds like the Hammond-Leffler postulate I teach in Organic Chemistry 1 – easy to implement, in principle. Fortunately enough, we had developed a docking program which included all the MM routines necessary for exhaustive conformational sampling and molecular optimization. Converting those into a small molecule conformational search and optimization program and including this Hammond-Leffler postulate led to the creation of Asymmetric Catalyst Evaluation (ACE 1.0, 2008).
As the years passed, we continued to develop ACE at a gradual pace as funding and my “spare” time allowed. We reported on an improved version in 2011. Although there were dozens of users of our drug discovery programs worldwide by this time, we only had one external user of ACE. This was partially a reflection of the contrast between the widespread adoption of computer-aided drug design on the one hand, and organic chemists’ enduring preference for DFT on the other. But usability was also a major barrier to a broader uptake of our programs.
Josh Pottel and I took this challenge on and developed Constructs, a program that could work with 2D catalysts and substrates drawn in any ChemDraw-like program and, provided a reaction template, to build a starting TS structure. A sketcher, dispensing with the need for a fancy graphical interface was also developed. But there remained a major problem: the reaction-specific template used by Constructs was difficult to build. Luckily enough, Mihai Patrascu joined the group with an objective to combine organic chemistry and QM. He developed Quemist and integrated it into Constructs. Additional tools, such as Finders and React2D for virtual combinatorial chemistry reported previously, were added, along with modular workflows, to enable chemists to simulate their daily life in the lab.
All that brings us to where we are today: a completely virtual lab which can search catalogs (Finders), virtually synthesize catalysts (React2D) and test catalysts for catalytic efficiency (Quemist) and stereoselectivity (Ace). Tasks can be assembled in modular workflows and large projects can be virtually designed and performed on a computer prior to any time-consuming wet lab work.
23 years after observing a mismatched case in carbohydrate derivatives, I am pleased to report Virtual Chemist. The circle is completed! Where to from here?
Mihai Burai Patrascu, Joshua Pottel, Sharon Pinus, Michelle Bezanson, Per-Ola Norrby, Nicolas Moitessier From desktop to benchtop with automated computational workflows for computer-aided design in asymmetric catalysis. Nature Catalysis, 2020, 3, 574-584




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in